Exerc Sport Sci Rev. 2012;40(4):195-203.
http://www.medscape.com/viewarticle/771738When compared with age-matched men, premenopausal women have lower mortality, which is often thought to be caused by the protective effects of estrogens.[30] For a variety of reasons, most women must deal with the onset of decreased estrogen concentrations at some point in their lives. Reductions in circulating estrogens are associated most commonly with the onset of age-induced menopause. In addition, numerous clinical reasons may cause a woman to face loss of estrogen function before natural menopause. For example, oophorectomy is used clinically in women as a prophylactic approach to reduce the risk of genetically screened estrogen-positive cancers or for alleviating symptoms of chronic migraine.[16] In addition, using estrogen receptor antagonists as a treatment for estrogen-positive cancers also can result in ovarian failure. Finally, a small percentage of young women develop primary ovarian insufficiency, leading to abnormally low levels of circulating estrogens.[16] In this review, we define the loss of "estrogen function" as reductions in tissue-specific estrogen signaling caused by significant losses in circulating estrogens and/or pharmacological inhibition of the estrogen receptors.
Unfortunately, decreases in circulating estrogens in women result in increased risk for a number of clinical health issues that can lead to increased mortality. For example, reduced estrogen levels result in significant alterations in skeletal muscle, bone, cardiovascular, and brain functions,[30] with clinicians often prescribing estrogen or hormone therapy (HT) as a means to prevent or attenuate the onset of these symptoms. With the recent release of results from the Women's Health Initiative (WHI), there has been a substantial decline in the clinical use of HT because a portion of the data suggested that HT increased the risk of breast cancer and stroke.[8] The broad sweeping conclusions of the WHI have since been questioned for multiple reasons,[30] but these questions have not curtailed the reduction in HT prescription. Thus, the WHI has had a clear impact on the use of HT by clinicians in addressing issues on women's health, and it is important that appropriate alternative approaches be provided for women.
An important goal for women's health research is to define and understand mechanisms by which estrogens impact cell function across a variety of tissues. In women, estrogens encourage physiological mechanisms that prevent chronic disease; however, the loss of estrogen function results in the development of pathological function, indicating that estrogens are a key regulator of the cellular phenotype. Thus, if we can define the estrogen-regulated mechanisms that affect tissue function, we may be able to develop interventions to prevent the development of chronic disease in women with reduced levels of circulating estrogens. The goal of this review is to highlight recent findings concerning changes in metabolic function in adipose and hepatic tissue under conditions of reduced estrogen levels. In addition, we will explore the hypothesis that exercise training is an intervention that should be used as a primary approach to prevent the onset of metabolic disease in women who are experiencing a loss or decrease in estrogens.
Adipose Tissue Expansion Under Conditions of Reduced Estrogen Function
In females, a reduction in estrogens results in significant increases in fat mass in the visceral region; however, this change does not always equate to increases in overall body weight. In this review, we will use the term "lipid" to refer to fatty acid–based molecules, and the term "fat" will be used in an anatomical sense to describe adipose tissue. The impact of estrogen on adipose tissue is evident when comparing anatomical fat storage in males with that in females. Males tend to store excess lipid in the abdominal region (i.e., visceral fat), an android distribution pattern, whereas females with normal cycling estrogen patterns preferentially store lipid in the lower half of the body, a gynoid pattern of fat distribution. However, when estrogen levels are reduced chronically in women, there is an increase in visceral fat mass that results in a shift in body shape from a gynoid to an android pattern. With this increase in visceral fat mass, women are at an increased risk for developing metabolic and cardiovascular diseases.[30] The ovary secretes numerous hormones, but evidence suggests that loss of estrogen is critical because of its powerful effect on visceral adipose tissue function. For example, the use of HT in women can attenuate increases in visceral fat mass observed in women after the menopausal transition.[30] In a similar fashion, chronic 17β-estradiol treatment in animal models with estrogen deficiencies can prevent or even reverse the development of visceral adiposity.[32]Increases in visceral adiposity are of serious concern because of the significant link that exists between visceral fat storage, the development of metabolic disease, and increased mortality. The anatomical location of fat is a critical consideration, in that excess lipid storage in different anatomical fat depots has different consequences on glucose disposal. Fat storage in subcutaneous depots located in the lower half of the body typically is not associated with changes in insulin sensitivity, whereas excess lipid storage in the visceral region is associated with increases in peripheral insulin resistance. Based on the changes observed in visceral fat mass in women with reduced estrogen levels, it is clear that ovarian hormones, likely estrogens, play a critical role in the regulation of lipid metabolism in females.
Using Animals to Understand Metabolic Changes Induced by Reduced Estrogen Function
From a physiological standpoint, human studies of estrogen function in liver and adipose tissue in females are both experimentally feasible and informative. However, mechanisms are often hard to identify in human studies because of limitations of studying tissue biopsies that are small in size and not readily available. Study design and overall conclusions also are often affected by large variations in patient/subject characteristics. These difficulties have led to the use of different animal models to study the role of the ovary or estrogens in the regulation of tissue function. The ovariectomy model (i.e., bilateral surgical removal of the ovaries (OVX)) is one of the most commonly used animal models to study the role of female sex steroids in tissue function.[6,9–11,14,21,24,26,31,32] After OVX, circulating estrogen levels are reduced by approximately 70% to 90%,[11,26] a reduction comparable to that observed in females after natural or surgically induced menopause. Although the OVX model is often criticized for the abrupt nature of the onset, it still has provided critical information concerning women's health. In addition, OVX is relevant clinically because numerous premenopausal women undergo the same ovariectomy (i.e., oophorectomy) surgery for a variety of clinical reasons. Thus, it is unacceptable to discount the scientific importance of the OVX model because of the limitations it has in mimicking every aspect of age-related menopause when one considers the number of women who undergo oophorectomy before the onset of age-induced menopause. In addition, a number of genetic models are available that can be used to study estrogen function, including the estrogen receptor–knockout (ERKO) mouse and the aromatase-knockout (ARKO) mouse.For unknown mechanistic reasons, there is a clear association of lost of estrogen function with the onset of metabolic disease. For example, the OVX model exhibits a significant increase in visceral adiposity (Table), which is associated with increased peripheral insulin resistance.[31,32] These changes in visceral adiposity and insulin resistance are recapitulated in both the ARKO and the ERKO-[alpha] mouse models, suggesting that, regardless of the chosen animal model, loss of estrogen function leads to the development of metabolic disease.[12,15] It is clear that loss of female sex steroid function results in an increased risk of developing metabolic abnormalities, indicating that estrogens likely protect women from acute metabolic insults.
Adipocyte Hypertrophy Under Conditions of Reduced Circulating Estrogens
In addition to the visibly apparent changes in adipose tissue mass, alterations in fat cell (adipocyte) function occur when estrogen levels are compromised chronically. Changes in adipocyte function are caused by altered morphological changes in the fat pad, as well as to changes in key regulators of fat storage and breakdown. Morphologically, adipose tissue expansion may occur via an increase in adipocyte size and/or an increase in adipocyte number. When expansion occurs because of an increase in adipocyte number, peripheral insulin sensitivity often is preserved; whereas in contrast, when expansion is caused by an increased adipocyte size, metabolic consequences, such as reduced peripheral insulin sensitivity, commonly are observed.[27] In humans, a link between ovarian hormone status and visceral adipocyte size has been reported, with ovarian hormone–deficient women exhibiting significantly greater visceral adipocyte size compared with women with intact ovarian function.[29] We have found that OVX animals exhibit substantial increases in visceral adipocyte size compared with ovary-intact female (SHAM) animals (Fig. 1). The increase in adipocyte size develops over time, with substantially larger adipocytes readily apparent 8 wk after the OVX surgery (Fig. 1), without significant changes in adipocyte number (Wohlers LW and Spangenburg EE, unpublished data, 2012). The initial increase in the size of the adipocyte is visible 2 wk after the OVX surgery, with continued hypertrophy of the adipocyte visible for the next 6 wk (Wohlers LW and Spangenburg EE, unpublished data, 2012). Similar findings have been observed at later time points by other laboratories.[6,24]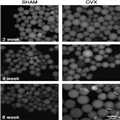
Figure 1.
Ovariectomized (OVX) mice exhibit larger visceral adipocytes at various time points after surgery (2, 4, or 8) compared with SHAM animals. The OVX surgery was performed at 8 to 10 weeks of age, with the time points referring to point of tissue collection after the surgery. Isolated adipocytes are stained with BODIPY 493/503 and imaged with fluorescence microscopy. The scale bar equals 100 µm.
Adipocyte Dysfunction Under Conditions of Reduced Estrogen Levels
Unfortunately, because of the experimental difficulties in obtaining visceral fat biopsies from women, the mechanistic study of visceral adipocyte function as they relate to estrogen function is challenging. However, through the use of animals, we have gained substantial insight into the relationship between estrogens and adipocyte biology. Under normal physiological conditions, excess circulating free fatty acids (FFA) are stored efficiently as triacylglycerol (TAG), and lipolytic breakdown of TAG is initiated by stimuli such as starvation or exercise. However, in certain disease states, the regulatory mechanisms of TAG metabolism are disrupted, contributing to the onset of various metabolic diseases. OVX animals exhibit significant elevations in basal lipolysis in visceral adipose tissue; however, the visceral fat pad in OVX animals does not respond appropriately to stimuli that are expected to induce lipolysis.[6,31,32] We have found in OVX animals that increases in visceral adipocyte size are associated with increased basal lipolytic activation. After OVX, mice exhibit increases in basal lipolytic rate, as indicated by both elevated circulating serum glycerol and FFA levels, which are mimicked in organ bath and isolated adipocyte measures as well.[6,32] In addition, the in vitro stimulated lipolytic response of adipocytes isolated from OVX animals is impaired but is rescued in mice supplemented with estrogen.[6] OVX mice also exhibit impaired lipolytic signaling activation in response to an acute bout of running, a physiological stimulus of lipolysis.[31] At this point, it is unclear why stimulated lipolysis in visceral fat is blunted in OVX mice, but it is possible that basal lipolysis is upregulated to such an extent that the tissue becomes unresponsive to further stimulation.The observed alterations in lipolytic dynamics when ovarian function is compromised are accompanied by changes in protein content of key regulators of adipocyte lipolytic function. Specifically, OVX animals exhibit significant increases in visceral adipose tissue glycerol lipase (ATGL) protein (Fig. 2) and an increase in the interaction between ATGL and comparative gene idenfication-58 (CGI-58) compared with SHAM animals.[32] CGI-58 is a known activator of ATGL, thus suggesting that increased ATGL activation is a likely contributor to the higher basal lipolytic rates in the OVX animals. We also found decreases in perilipin (PLIN1) protein content in the OVX mice compared with that in SHAM mice (Fig. 2). PLIN1 is a lipid droplet coating protein that regulates both lipolytic rate by preventing unstimulated lipolysis and also by facilitating activated lipolysis.[2] Adipose tissue from knockout mice of PLIN1 exhibits increases in basal lipolysis,[2] which is similar to results seen in the OVX model. Together, these data suggest that loss of PLIN1 contributes to dysregulation of lipolysis in the OVX animals compared with the intact SHAM group. Our data also suggest that loss of PLIN1 content is paralleled by upregulation of another lipid droplet coating protein termed PLIN2 (ADRP).[31] Although PLIN2 retains some similarities to PLIN1, it does not provide the protective effect of PLIN1 in preventing unstimulated lipolytic attack,[2] which may also explain the increase in basal lipolyticrate seen in the OVX mice. In addition, overexpression of PLIN2 results in enhanced lipid storage,[2] thus suggesting that it may play a role in the increased lipid storage in the OVX mouse, which results in adipocyte hypertrophy. Surprisingly, when we gave the OVX animals access to voluntary running wheels, we failed to prevent this dysregulation in lipolytic function; however, supplementation of the OVX mice with 17β-estradiol was completely effective at preventing these changes from developing.[32] Our data also suggest that activation of lipolytic signaling mechanisms was impaired in an untrained OVX group in response to an acute bout of treadmill exercise compared with the SHAM group.[31] In response to the exercise bout, there was lower HSL phosphorylation (Fig. 2), which was likely a consequence of reduced PLIN1 content. Serine phosphorylation of PLIN1 in the protein kinase A consensus sequence is necessary for maximal lipolysis, which coincides with phosphorylation of HSL and direct interaction of HSL with PLIN1 in the PAT domain.[2] However, PLIN2 appears to lack the ability to support maximal rates of lipolysis induced by HSL, thus the upregulation of PLIN2 likely contributes to altered lipolytic response in the OVX animals. When compared with the intact female animals, OVX animals exhibit significant changes in key lipolytic signaling proteins that likely contribute to increases in the basal lipolytic rate.
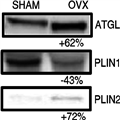
Figure 2.
Immunoblotting for key lipolytic proteins in visceral adipose tissue from ovariectomized (OVX) and SHAM animals. Below each example is the percent difference in means that was determined in our previous publication (data from (31)). ATGL, adipose tissue glycerol lipase; PLIN, perilipin.
The ability of the adipocyte to store lipid as TAG is limited, which was first demonstrated by the Czech lab that found significant down-regulation of fatty acid synthesis in large adipocytes.[5] The inability to store more lipid is referred to as the "overflow hypothesis," where the lipid droplet is unable to store more TAG, resulting in increased levels of FFA in circulation, an effect observed in the OVX animals. This loss of regulatory control of TAG dynamics would be expected to contribute to increased risk of developing lipotoxicity in peripheral tissues because of increased exposure to higher concentrations of circulating lipid. In the case of this review, lipotoxicity is defined as the excessive accumulation of cellular lipid that contributes to chronic cellular dysfunction. Indeed, we recently demonstrated that hepatic tissue is a likely target of this spillover because of the anatomical relationship of the liver with visceral tissue via the portal circulation.[14]
Changes in Lipid Metabolism in Hepatic Tissue Under Chronic Conditions of Reduced Estrogen
Significant decreases in circulating estrogens induce adipocyte dysfunction that systemically exposes peripheral tissue to chronic increases in circulating FFA. In the case of visceral fat, FFA are released into the portal circulation and are first directed to the liver. In fact, 59% of stored hepatic TAG is derived from FFA in the portal circulation, 26% from de novo lipogenesis, and 15% from the diet.[7] Thus, an increase in FFA from portal circulation exposure would increase the risk of hepatic lipotoxicity. The development of fatty liver through the association of visceral adiposity is often referred to as the "portal vein hypothesis." Studies show that FFA released through visceral adipose tissue lipolysis are the predominant sources of hepatic lipids in patients with nonalcoholic fatty liver disease (NAFLD).[7] The risk of hepatic lipotoxicity likely is exacerbated if metabolic mechanisms within the exposed hepatocytes are compromised, preventing the cell from tolerating the increased flux of FFA. Here, we are proposing a two-hit hypothesis in that estrogens provide a multifactorial means for regulating metabolic function in women and that the loss of estrogens leads first to adipocyte dysfunction and second to a loss of mechanisms that would allow peripheral tissue to tolerate increases in lipid exposure. In the following paragraphs, we discuss data demonstrating that decreases in estrogens in women leads to metabolic dysfunction in hepatic cells that potentially could contribute to an increased risk for developing diabetes.Women are not only inclined to the development of central obesity after menopause, but they also are susceptible to lipid accumulation in the liver. Obesity is often associated with the development of NAFLD.[22] NAFLD is the result of excess TAG storage in the liver, comprising a histological spectrum ranging from simple hepatic steatosis to advanced fibrosis and cirrhosis.[22] Intrinsically linked to the significant increases in lipid accumulation in the liver is a significant increase in the risk for developing insulin resistance and, ultimately, metabolic syndrome. Premenopausal women are at lower risk for developing NAFLD than age-matched men; however, when women become postmenopausal, there is a significant increase in risk for developing NAFLD.[17,28] Estrogens appear to protect against the development of NAFLD even under conditions (i.e., high-fat diet) that would accentuate lipid accumulation in the liver (Fig. 3).[4] As discussed above, we and others have shown that increases in basal lipolysis in OVX mice result in enhanced FFA release from the visceral depot likely into the portal vein, ultimately leading to increased lipid storage because of increased exposure of the hepatic tissue.[6,14,24,32] Thus, the explanation for the increased risk of NAFLD development in women under conditions of reduced estrogen levels is likely multifactorial.
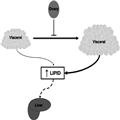
Figure 3.
Endocrine function of the ovary prevents excess adipose tissue expansion in the visceral region and release of excess free fatty acids (FFA) into the portal circulation.
Increases in hepatic lipid (i.e., TAG) content can be the result of alterations in a number of metabolic events. For example, increased TAG content can be a direct result of increased FFA influx or caused by increased synthesis of fatty acids via fatty acid synthase (FAS). In addition, TAG accumulation could result from decreased export of TAG via very low density lipoprotein or caused by decreased mitochondrial oxidation. In other words, a host of factors can contribute to changes in hepatic TAG content, and dysfunction of any of them can lead to significant increases in hepatic TAG content. In the OVX animals, significant increases in hepatic TAG content are often seen in the rat model. However, the data in the mouse OVX model are more equivocal, in that in the mouse, there is an increase in stored TAG; however, the increase does not reach the same magnitude as that in the rat.[14,21] What is critical to consider is that these increases in stored hepatic TAG are occurring even though the rodents are being maintained on a relatively low-fat diet. If the diet is changed to a high-fat diet, both mice and rats develop severe forms of NAFLD, indicating that the loss of estrogen exposure leads to a significant hypersusceptibility to NAFLD, which is often seen when fat exposure is increased. An additional complication to consider is the susceptibility of different strains of rodents to metabolic insults, which adds another layer of complexity to the interpretation of the published data. At this point, mechanisms within the hepatic tissue remain poorly defined, and only a few investigations have provided data to indicate potential mechanisms for why loss of estrogen might lead to increased susceptibility to NAFLD.
In our hands, we have found little indication of OVX mice increasing hepatic lipid storage because of increases in FAS or acetyl-Co carboxylase (ACC) protein content.[14] In contrast, D'Eon et al.[6] found significant decreases in hepatic FAS and ACC messenger ribonucleic acid (mRNA) expression in response to exogenous 17β-estradiol delivery to the OVX mouse, suggesting that estrogens may negatively regulate de novo synthesis of FA. However, it should be noted that the study design by D'Eon et al.[6] did not contain a SHAM control group, so it is impossible to determine if the OVX group had elevated levels of hepatic mRNA levels of FAS and ACC. In a follow-up study, Rogers et al.[24] found increased mRNA levels of hepatic FAS and ACC, coupled with histological indices of hepatic steatosis. The differences with our work are likely caused by the time of tissue collection, in that in our study, we collected tissue 8 weeks after OVX, whereas Rogers et al.[24] collected 10 weeks after surgery. Furthermore, OVX rats exhibit significant increases in hepatic ACC mRNA expression compared with intact cycling females.[21] Whereas further investigations are necessary to elucidate the mechanisms that link OVX to alterations in hepatic tissue, it appears that the level of activation of the FAS pathway may influence the severity of TAG accumulation in the OVX model and likely result in the development of NAFLD.
Decreases in Circulating Estrogens Leads to Increased Hepatic Stearoyl CoA Desaturase Activity
Although we did not find large increases in total TAG content, we did find significant changes in the composition of the fatty acids stored as TAG in the liver.[14] Significant changes in the composition of stored fatty acids can alter numerous physiological processes including signal transduction, insulin sensitivity, tissue metabolic rate, and fatty acid storage kinetics.[20] Specifically, we found a significant increase in the percentage of fatty acids in the monounsaturated form (16:1; 18:1) in the OVX mouse when compared with that in the SHAM mouse. Calculation of the desaturase index of the fatty acids stored as TAG in the liver demonstrated that there was a significantly greater amount of monounsaturated fatty acids (MUFA) than saturated fatty acids stored in OVX mice when compared with that in SHAM mice (Fig. 4). In the liver, the conversion of a saturated fatty acid (16:0, 18:0) to a MUFA (16:1, 18:1) is regulated enzymatically by the enzyme stearoyl CoA desaturase (SCD-1). In addition, we found a significant increase in the hepatic SCD-1 protein content in the OVX mice compared with that in SHAM mice (Fig. 4),[14] which also has been found by others at the mRNA and protein levels.[34] This finding was informative because MUFA are stored preferentially, as opposed to saturated fatty acids that are often favored for oxidation by the mitochondria.[20] SCD-1 is localized at the endoplasmic reticulum and plays a critical role in regulating cellular function across a variety of tissues; however, hyperactivation of SCD-1 can contribute to the development of metabolic disorders. Specifically, mice with global tissue deletion of SCD-1 conserve peripheral insulin sensitivity even while on a high-fat or high-carbohydrate diet (for detailed review, see).[20] In our hands, OVX mice develop visceral adiposity, glucose intolerance, and dysregulation of metabolic function in hepatic tissue while on a normal chow diet (70% carbohydrate; 10% fat, and 10% protein); thus, it is not unreasonable to suggest that hepatic SCD-1 may be a critical mediator of metabolic dysfunction in OVX mice. Finally, we found that SCD-1 content was correlated positively with blood glucose levels in SHAM and OVX mice (Fig. 4), suggesting that in the OVX model, there is a potential relationship between the increase in hepatic SCD-1 levels and the onset of glucose intolerance. These data conceptually provide evidence to our two-hit hypothesis idea in that the combination of events, visceral adipocyte dysfunction and increased hepatic SCD-1 content, would make individuals more susceptible to NAFLD and the development of overt metabolic disease.
Figure 4.
A. Sedentary ovariectomized (OVX) animals exhibited significant increases in the hepatic desaturase index (C18:1/C18:0) of extracted triacylglycerol (TAG) compared with SHAM animals, whereas exercise prevented any changes in the desaturase index in the OVX animals. †Statistically different from all other groups; *statistically different from sedentary SHAM (P < 0.05). B. Sedentary OVX animals exhibited significant increases in hepatic stearoyl CoA desaturase (SCD-1) content compared with all other groups. Exercise prevented the increase in hepatic SCD-1 content of the OVX mice. A representative blot of SCD-1 is shown beneath the graph. C. Hepatic SCD-1 protein content is correlated positively with blood glucose levels (4Y5 hours' fast) in sedentary OVX (
 ) and SHAM (
) and SHAM (
 ) animals. Data taken from our previous work (14).
) animals. Data taken from our previous work (14).
Chicken or the Egg: Why is Metabolic Function in the Liver Altered?
Decreases in estrogen function that are induced by surgical, genetic, or pharmacological means lead to complex phenotypes throughout the animal. Specifically, because of the number of estrogen sensitive tissues, loss of estrogen signaling throughout the entire body leads to numerous pathological changes (i.e., obesity, insulin resistance, and physical inactivity) that often complicate the interpretation of the resulting data. It is often impossible to truly discern secondary effects versus primary effects of decreased estrogen function. For example, we have shown previously that OVX rats recover poorly from a bout of muscle atrophy when compared with intact females;[26] however, is this result caused by effects of estrogens specifically on the muscle, or is it complicated by the fact that the animals are obese and insulin resistant? Interestingly, changes found in the OVX, ERKO, and ARKO mice are similar qualitatively, thus, indicating that, regardless of how estrogen signaling is reduced, it results in substantial, but similar, physiological or metabolic deficiencies. Furthermore, exogenous delivery of 17β-estradiol can often prevent or reverse the metabolic dysfunction in the OVX and ARKO mice, suggesting the estrogens likely are the critical regulator and not another ovarian hormone. However, it should be noted that the delivery approaches generally are not physiological because they result in chronic doses of 17β-estradiol that are often on the upper levels of physiological concentrations rather than cyclic exposure of varying 17β-estradiol levels seen in females. The challenge remains for investigators to identify which effects are specific to the role estrogen plays in that tissue and which are secondary effects. Recently, two laboratories have shown elegantly that metabolic disruptions develop through specific genetic ablation of the estrogen receptor from myeloid cells or neurons within the hypothalamus showing the importance of tissue-specific estrogen signaling.[23,33] At this time, what is clear is that lost estrogen function leads to increased risk of numerous chronic health conditions, all of which are important critically to women's health; however, what is often not clear is which effects primarily are caused by lost estrogen function and which are secondary effects caused by changes elsewhere in the body.Our data in the OVX indicate that SCD-1 activation is higher under conditions of reduced estrogen concentrations (Fig. 4).[14] Similar results have been achieved in the ERKO mouse where SCD-1 mRNA expression was significantly elevated when compared with the wild-type mice, which was coupled with disruptions in glucose homeostasis.[3] Importantly, in a follow-up study, it was identified that estrogens negatively regulate the SCD-1 promoter, implicating that the loss of estrogens likely contributes to increased SCD-1 activation independent of obesity.[4] Albeit, it is very possible that development of the visceral adiposity additively increases SCD-1 promoter activation because the promoter is enhanced with FFA exposure,[13] again providing further evidence to the complexity of this phenotype. Regardless, treatment of obese animal models with 17β-estradiol or with estrogen receptor agonists attenuates or reverses the metabolic abnormalities induced by a variety of metabolic insults.[4,6,10] As a whole, the data clearly suggest that estrogens play a protective effect in preventing metabolic disease. Unfortunately, because of the complexity of the developing phenotype, it typically is unclear which targets truly are estrogen sensitive. It is critical that research continues to examine the mechanisms behind how estrogens contribute to the regulation of tissue function to determine where metabolic defects will develop during conditions of estrogen deficiency. However, it should be noted that if approaches are to be developed to prevent or reverse the effects of estrogen dysfunction they should be tested on clinically relevant models such as the OVX model and not just limited to genetically manipulated animal models. Ultimately, this will be critical because the WHI has resulted in a significant reduction in the use of the estrogen therapy as a means to treat women experiencing physiological changes caused by estrogen deficiency.
Increasing Physical Activity in OVX Mice Prevents Changes in Hepatic Tissue
In an effort to determine if the changes in hepatic SCD-1 protein content were preventable, we provided the mice with access to voluntary running wheels. We found that access to wheels resulted in attenuated increases in visceral mass and changes in hepatic TAG content in both the intact female mice and the OVX mice.[14,32] It should be noted that the intact control female mice ran a significantly greater amount of distance each night (~8200 km every 24 h) compared with the OVX mice (~2400 km every 24 h), which suggests that it only was necessary to slightly increase the activity levels to prevent the TAG accumulation. This is in agreement with others who found that treadmill running in OVX rats results in significant reductions in hepatic TAG content compared with sedentary groups.[21] Our evidence also suggested that increased physical activity caused by the wheel running resulted in decreased hepatic expression of diacylglycerolacyltransferase (DGAT1); however, the decrease was similar in the intact females and OVX mice. The decrease in DGAT1 is intriguing because DGAT1 represents the final regulatory step in the assembly of a TAG and may suggest that DGAT is sensitive to changes in activity levels. Barsalani et al.[1] demonstrated that treadmill running in OVX rats attenuated increases in DGAT2 compared with the OVX sedentary group. In addition, we indicated earlier that the FAS pathway may contribute to TAG accumulation in the liver. We found no effect of voluntary wheel running on hepatic FAS or ACC protein content in the OVX or intact female mice; however, others have found that forced treadmill running of OVX rats prevented increases in ACC mRNA expression.[21] Although the mechanism induced by exercise to prevent the hepatic TAG accumulation remains to be determined completely, it is clear that, like other models of obesity,[22] OVX rodents do not accumulate hepatic TAG significantly when they are given the opportunity to exercise.Wheel running also prevented the increase in hepatic 16:1 and 18:1 content of the OVX mice, which was associated with a lower desaturase index and reduced SCD-1 content when compared with those of the sedentary OVX mice (Fig. 4).[14] This finding is in agreement with that of others who found that in the OVX rat forced treadmill running prevented increases in hepatic SCD-1 mRNA and protein content when compared with the OVX sedentary group.[34] These data provide critical information indicating that loss of estrogenic function leads to increased SCD-1 activity that can be prevented by increasing the physical activity levels of the animal.
This would suggest that even small increases in wheel running completely prevented the metabolic changes in the OVX rodent, resulting in maintenance in hepatic phenotype that is similar qualitatively to the sedentary intact female mice. Furthermore, as suggested by others,[21] providing OVX rodents with the ability to exercise through wheel running or treadmill activity induces estrogen-like effects that induce a more favorable metabolic phenotype. However, it should be noted that the cellular mechanisms induced by exercise and estrogens likely are different. In addition, an important consideration is that decreases in the physical activity levels of the OVX model likely exacerbates the development of the metabolic disruptions seen within the animal.
Physical Activity is a Potential Intervention for Treating Women With Reduced Estrogen Function
If estrogen therapy is not considered a safe intervention, then it absolutely is critical that clinicians consider alternative approaches. The data are clear and compelling that decreases in estrogen levels or estrogen receptor function result in significant increases in risk for developing metabolic disease in women. When examining the metabolic changes induced by estrogen dysfunction, it is apparent why exercise or increases in the levels of physical activity would be so effective as an intervention. Loss of estrogen function leads to insulin resistance,[10] visceral adiposity,[31,32] hepatic dysfunction ([14,17]), and striated muscle dysfunction;[19,26] however, every one of these alterations can be prevented or reversed by increasing physical activity levels. Although the magnitude of studies done on women under conditions of reduced estrogen function is relatively low, there is indication that exercise training is an effective approach. For example, Ryan et al.[25] found that weight loss alone did not improve glucose utilization and insulin sensitivity in postmenopausal women; however, when weight loss was coupled with exercise training, it resulted in a significant improvement in both outcomes. Similar results were shown by You et al.,[35] who demonstrated that diet alone was not effective at reducing circulating inflammatory markers in postmenopausal women; however, when coupled with exercise training, there was a significant reduction in these same cytokines. These data suggest that exercise training is an intervention that is critical to women who clinically are experiencing reductions in estrogen function. Also, it is possible to extend beyond just metabolic dysfunction because recent data have shown that physical activity improves the survival rates of postmenopausal women with breast cancer.[18] In a qualitative fashion, the data indicate that longer duration bouts of exercise provide the most benefit; however, the intensity of activity did not seem to be critical.[18] Interestingly, we can extrapolate these findings to ours in that we found that very low levels of physical activity were effective in preventing the development metabolic disruptions in the hepatic tissue.[14] The ability of low-intensity exercise to be effective is critical because reductions in circulating estrogen levels are associated with reduced muscle force production[19] and, thus, a loss of muscle strength likely would hamper the individual from completing high-intensity exercise bouts. With both mechanistic and epidemiologic-based evidence demonstrating the importance of using physical activity as a means to improve health outcomes of women under conditions of reduced estrogen function, it seems that it will be necessary to analyze critically and determine scientifically appropriate approaches for using exercise as a therapeutic intervention in women.Overall Conclusions
The broader implication of these data is that physical activity may be an effective substitution for estrogen therapy in women, but it is unclear if physical activity as a clinical intervention is used in women who are experiencing reduced estrogen function. In addition, we feel that if the WHI is indicating that HT may not be a safe approach for women, then efforts must be made to define and promote alternative approaches for women. Thus, a critical next step would be conduction of more detailed human studies to determine the efficacy of using exercise training to prevent metabolic dysfunction in women who are experiencing reduced levels of estrogen or reduced estrogen receptor function. Considerations that need to be addressed would be the amount of necessary activity, intensity, and so on. Furthermore, we would predict that if loss of estrogenic function is caught early in women, then actual intervention might require only low levels of physical activity to prevent the accumulation of the metabolic defects. Conversely, we also suggest that it likely is more difficult (i.e., requires more strenuous exercise) to reverse a metabolic defect with exercise training, thus indicating that early screening by clinicians would be a critical aspect to consider. Overall, the numerous published data points indicate that we need to think carefully about the influence of sex steroids on tissue function and the interaction that appears to exist with physical activity.http://www.medscape.com/viewarticle/771738